ブログ
医薬品のパテントリンケージ制度の概要
2022.08.10
SHARE![]()
![]()
![]()
はじめに
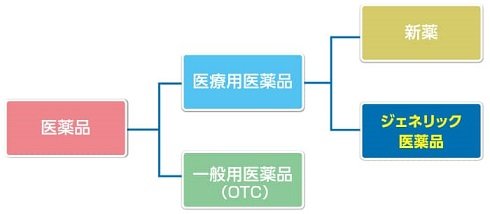
医療用医薬品には、新薬と後発医薬品(ジェネリック医薬品)があります。新薬は、既に承認を与えられている医薬品と有効成分、効能・効果等が明らかに異なる医薬品であり、通常、特許により保護されています。本稿では、特許で保護されている新薬を先発医薬品といいます。後発医薬品は、先発医薬品の特許が切れた後に先発医薬品と同じ有効成分を同じ量で作られた、効き目も安全性も同等である医薬品です。
(出所:日本ジェネリック製薬協会|ジェネリック医薬品とは (jga.gr.jp))
先発医薬品は、一般に、化合物スクリーニングや誘導化等を行う創薬研究、試験管中の実験や動物対象の実験を行う非臨床試験、ヒトを対象とする臨床試験、製造販売のための薬事当局への承認申請・審査という長い道のりを経て発売されます。

(参考:創薬と育薬 | 一般の方向けおくすりガイド | 日本製薬工業協会 (jpma.or.jp) )
先発医薬品に係る特許には、先発医薬品の有効成分を保護する物質特許、有効成分の効能・効果を特定した用途特許(医薬品として承認されている適応症(用途)を保護する特許)、先発医薬品の添加剤等が特定された製剤特許、先発医薬品の有効成分に関する製法特許等がございます。
これらの特許に係る発明は、先発医薬品の研究開発の各段階別に続々生まれることもあり、また先発医薬品の独占販売期間を延ばすというライフサイクルマネジメント戦略の観点から、先発医薬品メーカーは通常何段階に分けて特許出願をしております。その結果、有効成分の物質特許や効能・効果を特定した用途特許の存続期間が満了した後も、添加剤や組成が特定された製剤特許等が有効に存続する場合がございます。
医薬品のパテントリンケージ(Patent Linkage)制度とは
後発医薬品の販売後に、先発医薬品に係る特許の侵害訴訟等により後発医薬品の安定供給の問題が生じることのないよう、薬事当局が後発医薬品の承認にあたって、先発医薬品に係る有効特許を考慮する仕組みであり、後発医薬品の承認を先発医薬品の特許と連携させる制度です。
米国で始まった制度であり、米国では1984年に制定された"Drug Price Competition and Patent Term Restoration Act of 1984"という法律により導入されております。米国の制度は、①先発医薬品に係る特許情報の公開、②後発医薬品の承認申請行為を先発医薬品特許権者への通知、及び③当事者同士の紛争解決を柱としたものとなっております。
現在多くの国では米国の制度を模した米国型制度を実施しております。例えば、カナダ、オーストラリア、韓国等の国では、米国との自由貿易協定(FTA)により導入されており、中国も米国と締結した第1段階の経済・貿易協定(CHAPTER 1 Section C Article 1.11 「Effective Mechanism for Early Resolution of Patent Disputes」)によって導入され、昨年(2021年)6月より施行の改正特許法に盛り込まれております。
一方、欧州ではパテントリンケージ制度を実施しておらず、日本は後述する米国型制度とは異なるユニークな制度を実施しております。
米国の制度と中国の制度の比較
上述しましたように、中国も米国の影響を受けて導入されていますので、米国の制度と同じく、上述しました①~③を柱としますが、以下の点で多少異なります。なお、中国のパテントリンケージ制度の関連法規や具体的運用に関しては下記リンク先の記事に纏められております。
(記事:中国の医薬品のパテントリンケージ制度chizaiprism202112.pdf (tmi.gr.jp) )
まず、下記表に示す通り、当該制度の柱①に関しては、制度の対象とする医薬品の種類が米国と中国で異なります。米国では化学医薬品のみが対象となっているものの、中国では、化学医薬品だけではなく、バイオ医薬品及び漢方医薬品も対象としております。
|
|
米国 |
中国 |
|
|
柱① 先発医薬品に係る特許情報の公開 |
特許情報の登録・公開のDB |
オレンジブック(Orange Book Database) |
中国上市薬品の特許情報登録プラットフォーム |
|
適用対象 |
化学医薬品 ①有効成分の物質特許 |
・化学医薬品 ①有効成分の物質特許 |
|
|
バイオ医薬品については、パープルブック(Purple Book)にその関連特許情報を掲載し、漢方医薬品については相当する制度がないようである |
・バイオ医薬品 ①有効成分の配列特許 |
||
|
・漢方医薬品 ①組成物特許 |
|||
当該制度の柱②及び柱③に関しては、米中共に、後発医薬品メーカーが承認申請の際に宣言した第IV段乃至第4類の宣言内容やその根拠等を先発医薬品特許権者に通知することにより、後発医薬品の承認申請行為を先発医薬品特許権者へ告知する点では一致します。
ただし、第IV段乃至第4類宣言をした後発医薬品の承認申請者又は先発医薬品特許権者が、所定の期間内に訴訟等を提起した場合は、薬事当局がその後発医薬品の薬事承認を控える待ち(Stay)期間が設けられていますが、中国のほうが米国より短く規定されております。また、中国では、後発医薬品が先発医薬品に係る特許発明の技術的範囲への属否を争う手段として、次の項目でご紹介の事例のように、裁判所に訴訟を提起するルートと、中国特許庁に行政裁決を提起するルートが設けられており、後者は中国の独特の運用です。
|
|
米国 |
中国 |
||
|
柱② 後発医薬品の承認申請行為を先発医薬品特許権者への通知 |
第I段:オレンジブックに特許情報が提出されていないこと(Paragraph I Certification) |
第1類:特許情報プラットフォームに先発医薬品に係る特許情報が登録されていないこと |
||
|
第II段:その特許がすでに期限が切れていること(Paragraph II Certification) |
第2類:その登録されている特許が既に消滅又は無効とされたこと |
|||
|
第III段:その特許の有効期限が切れる日付を明記し特許の有効期限が切れるまで後発医薬品が市場に出回らないこと(Paragraph III Certification) |
第3類:その登録されている先発医薬品に係る特許の存続期間満了前に、薬事承認を申請した後発医薬品を発売しないこと |
|||
|
第IV段:その特許が無効である、またはその特許を侵害しないこと(Paragraph IV Certification) |
第4類:その登録されている先発医薬品に係る特許が無効とされるべきである、または後発医薬品がその特許発明の技術的範囲に属しないこと |
|||
|
柱③ 当事者同士の紛争解決 |
パテント・チャレンジ |
第IV段乃至第4類宣言をした後発医薬品の承認申請者と先発医薬品特許権者を当事者とする特許紛争 |
||
|
提訴乃至申立期間 |
45日 |
|||
|
紛争解決ルート |
特許権侵害要否の判断:司法ルート |
特許発明の技術的範囲への属否の判断:司法ルート、行政ルート(中国特許庁に行政裁決を提起するルート) |
||
|
特許の有効性の判断:行政ルート |
特許の有効性の判断:行政ルート |
|||
|
薬事当局の待ち期間 |
30ヶ月 |
9ヶ月(化学医薬品のみを対象とする) |
||
米国型制度のもう一つの特徴として、最初に販売承認を取得し、かつ、パテント・チャレンジに成功した後発医薬品メーカーに対して、後発医薬品の市場参入を促進させたことに対するインセンティブとして、後発医薬品の市場独占(Exclusivity)期間を与えることです。中国は、米国よりかなり長い独占期間を与えております。なお、米国型制度を施行している国でも、自国の製薬産業の環境に合わせ、例えばカナダ等では当該独占期間が設けられておりません。
|
|
米国 |
中国 |
|
後発医薬品の独占期間 |
180日 |
12ヶ月 |
中国の事例
上述しましたように、中国では昨年(2021年)6月より施行されている改正特許法にてパテントリンケージ制度が導入されており、同年7月に中国薬事当局、中国特許庁、中国最高裁よりそれぞれ公表された法令の施行に伴い、正式に実施しております。
今年(2022年)4月に、初めての訴訟一審事件と行政裁決事件が発生しました。
|
・訴訟一審事件:日本製薬大手の中外製薬株式会社 v 中国製薬大手の温州海鶴薬業会社 ・行政裁決事件:米国製薬大手のパーデュー・ファーマ(Purdue Pharma)社 v 中国製薬大手の宜昌人福薬業会社 |
ただし、いずれの事件においても製剤特許がパテントリンケージ制度における対象特許となりましたが、承認申請した後発医薬品がそれら先発医薬品の製剤特許発明の技術的範囲に属しないと判断されております。医薬品に一般に使用する添加剤はそれ自身が体に作用したり有効成分の治療を妨げたりするものではないため、後発医薬品に先発医薬品と異なる添加剤乃至異なる用量での添加剤を使用することができ、医薬品メーカーによってお薬を飲みやすい形や大きさに変える等の工夫が図られていますので、そのような後発医薬品は先発医薬品の製剤特許と異なる場合があります。
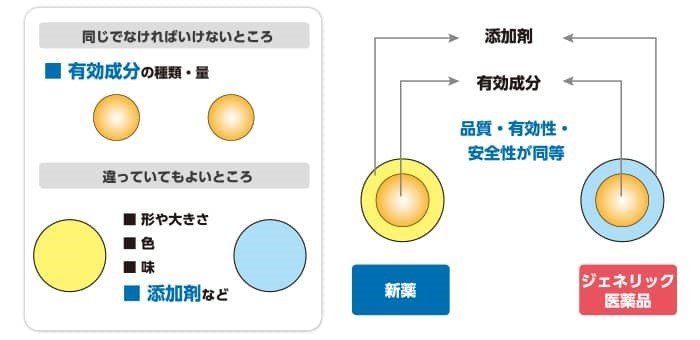
(出所:日本ジェネリック製薬協会|ジェネリック医薬品とは (jga.gr.jp) )
日本の制度
日本には、米国型制度のように、①先発医薬品関連の特許情報の公開や②先発医薬品特許権者等に対する後発医薬品の申請情報の通知に相当する制度は存在しませんが、日本なりのユニークな制度を実施しております。
日本では、法律上の規定は存在しなく、「医療用後発医薬品の薬事法上の承認審査及び薬価収載に係る医薬品特許の取扱いについて(平成21年6月5日厚生労働省医政局経済課長・医薬食品局審査管理課長通知)」及び「承認審査に係る医薬品特許情報の取扱いについて(平成6年10月4日薬審第762号審査課長通知)」に基づく厚生労働省の指導により、行政の実務として対処されております。
厚生労働省は、先発医薬品メーカーから報告された先発医薬品に係る「医薬品特許情報報告票」に基づいて、先発医薬品の有効成分の物質特許及び用途特許が有効に存続する場合には、原則後発医薬品の製造販売の承認を行いません。
また、後発医薬品の承認後、薬価収載の前に、先発及び後発医薬品メーカー間で、承認された後発医薬品について特許上の問題がないかを調整することとなっております。ただし、事前調整が妥結しないで終了する場合も生じており、調整不調で終わった場合も、後発医薬品メーカーから薬価収載の申請があれば、安定供給が可能と思われる品目についてその薬価収載を認めるという実状です。

このように日本では米国型制度と異なる制度を実施しております。TPP協定の交渉当時、米国の主張によりパテントリンケージ制度が提案され、TPP協定国である日本も米国型制度の導入を検討しましたが、米国のように訴訟が頻発することが懸念される等の理由で、先発医薬品メーカーと後発医薬品メーカーのいずれも反対する立場だったということです。結局2018年に発効したTPP11協定の第18.53条「特定の医薬品の販売に関する措置」に係る規定において、米国型制度と日本型制度の両方が盛り込まれております。
むすび
医薬品のパテントリンケージ制度は、国民の健康や生命に直接関わる医薬品に関して、後発医薬品の安定供給と先発医薬品の特許権による保護という両面を調整する制度ですので、公益に大きな影響を与える重要な制度と考えます。日本と米国ではそれぞれ自国の医療状況や製薬産業の実情等に基づいて上記のような日本型制度と米国型制度に設計されております。一方で、中国では米国の影響を受けて導入されている側面もありますので、今後、運用等を含め、自国の状況に合う制度にどのようにカスタマイズされていくのかに注目していきたいと考えます。また、中国でのパテントリンケージ制度に関し、有益な新たな情報がありましたら、本ブログで紹介していきたいと考えております。
Member
PROFILE

SHARE![]()
![]()
![]()