ブログ
TMI Blog: Healthcare & Pharmaceutical Regulations in Japan “Pharmaceuticals”
2026.03.03
SHARE![]()
![]()
![]()
What Are Pharmaceuticals?
In simple terms, “Pharmaceuticals” under the Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices (“PMD Act”) mean items which are intended to be used for the following purposes:
(i) Diagnosis, treatment or prevention of human or animal diseases; or
(ii) Affecting the structure and functioning of a human or animal’s body.
Pharmaceuticals are generally and broadly classified into “ethical drugs” which are intended to be used by physicians/dentists or under their prescriptions or instructions, in principle, and “OTC Drugs” which can be purchased by the general public without prescription and used at the person’s own discretion. OTC Drugs can be classified into “Drugs Requiring Guidance” and “General OTC Drugs”. Both Drugs Requiring Guidance and OTC Drugs are available at pharmacies or drugstores without prescription, but Drugs Requiring Guidance (such as Pharmaceuticals that have just switched from ethical drugs to OTC Drugs) require a pharmacist to provide required information and instruction in person using materials describing prescribed matters for proper use of the Drugs Requiring Guidance.
General OTC Drugs are further classified into Schedule I Drugs, Schedule II Drugs, and Schedule III Drugs depending on their respective risk.
The types of Pharmaceutical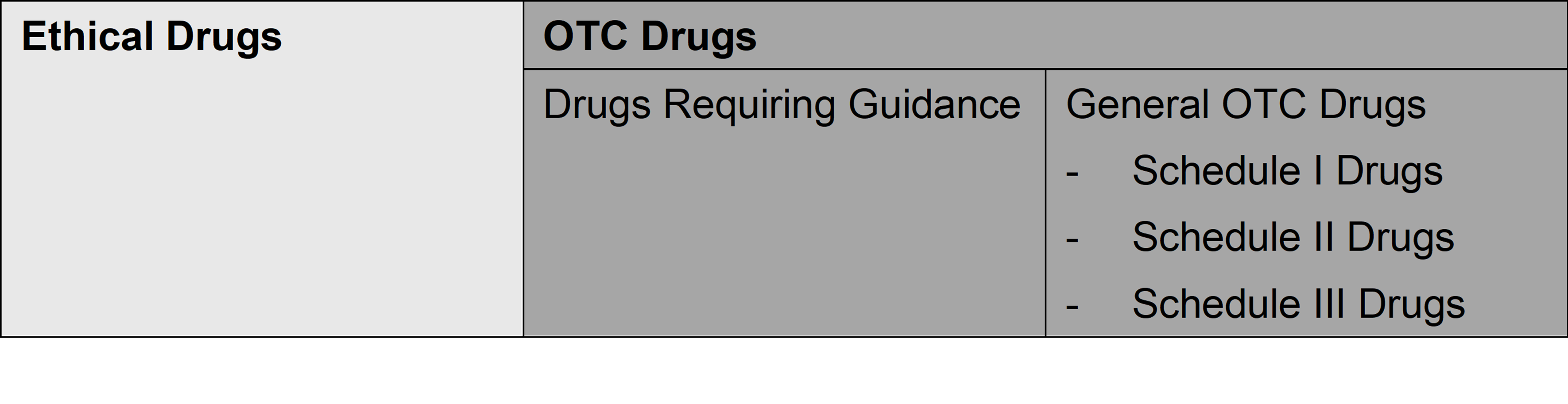
What Licenses and Approvals Are Required in Relation to Marketing of Pharmaceuticals?
The major necessary licenses and approvals are as follows:
(i) Marketing approval: Required for each Pharmaceutical on a product-by-product basis - please see 2-3 for details.
(ii) Marketing business license: Required to obtain marketing approval and to place Pharmaceuticals on the market - please see 2-5 for details.
(iii) Manufacturing license (or registration or accreditation (for foreign manufacturers), as the case may be): Required to manufacture Pharmaceuticals - please see 2-6 for details.
(iv) Sales license: Required to sell Pharmaceuticals - please see 2-7 for details.
What is “Marketing Approval” and Its Requirements?
Marketing approval (known as “Shonin”) is an authorization granted on a product-by-product basis; however, for certain Pharmaceuticals designated by the Minister of Health, Labour and Welfare (“Minister”), only a marketing notification is required. The Ministry of Health, Labour and Welfare is referred to herein as “MHLW”. Marketing approval needs to be obtained for each Pharmaceutical before the Pharmaceutical is placed on the market. Marketing approval is granted when the Pharmaceutical is determined as appropriate after the Pharmaceuticals and Medical Devices Agency’s (“PMDA”) or the relevant prefectural authority’s examination on its name, components, quantity, dosage, administration, efficacy, effects, side effects and other matters concerning the quality, efficacy and safety of the Pharmaceutical. The person or entity obtaining the marketing approval for the Pharmaceutical is called the “MAH (marketing approval holder)”. The MAH is ultimately responsible for the Pharmaceutical on the market.
The major high-level requirements for obtaining marketing approval are as follows:
- The applicant must have a marketing business license corresponding to the category of the Pharmaceutical that is subject to the marketing approval application.
- The manufacturing sites that manufacture the Pharmaceutical that is subject to the marketing approval application must have the corresponding manufacturing license, accreditation or registration.
- Manufacturing and quality control methods at the manufacturing sites must comply with GMP standards.
- The Pharmaceutical must demonstrate the claimed efficacy and effects.
- The Pharmaceutical must have demonstrable value as a drug without posing significant harmful effects that are disproportionate to its efficacy and intended effects.
An application for marketing approval must be submitted to the PMDA (or, for certain Pharmaceuticals, the relevant prefectural authority) together with the required supporting documentation and materials, including clinical trial results. Among the supporting documentation and materials for marketing approval, nonclinical study results related to drug safety must be collected and prepared in accordance with GLP*1, and clinical study results must be collected and prepared in accordance with GCP*2.
[*1 Ministerial Ordinance on Standards for the Conduct of Nonclinical Studies on the Safety of Pharmaceuticals.]
[*2 Ministerial Ordinance on Standards for the Conduct of Clinical Trials of Pharmaceuticals.]
Can a Foreign Company Obtain Marketing Approval?
Yes, but the foreign company needs to appoint a designated marketing approval holder in Japan (“dMAH”) to obtain the marketing approval. Specifically speaking, in order to obtain marketing approval for a Pharmaceutical, the applicant must have a marketing business license. A foreign company which is not located in Japan, however, cannot obtain a marketing business license. As an exception to such rule, the PMD Act allows a foreign company to obtain marketing approval by appointing a marketing business license holder in Japan (i.e., the dMAH) who will market the Pharmaceutical on behalf of such foreign company (such foreign company which holds marketing approval by appointing the dMAH, is the “Special Foreign Approval Holder”).
What is a “Marketing Business License” and Its Requirements?
A marketing business license is required for the “marketing” of Pharmaceuticals. For Pharmaceuticals, “marketing” means sales or provision of the items which are manufactured by the MAH itself, including those manufactured through contract manufacturing, or imported by the MAH itself. There are two types of marketing business licenses: (i) a first-class marketing business license required for prescription-only Pharmaceuticals and (ii) a second-class marketing business license required for Pharmaceuticals other than prescription-only Pharmaceuticals.
The major high-level requirements for a marketing business license are as follows:
- The applicant does not fall under any disqualification prescribed under the PMD Act.
- The quality control system adopted by the applicant must comply with GQP*3.
- The post-marketing safety control system adopted by the applicant must comply with GVP*4.
- The following personnel are appointed (as full-time employees, in principle) (known as “Sanyaku”):
- General Marketing Manager who is responsible for quality and post-marketing safety control (he/she must be a pharmacist, in principle and meet other requirements under the PMD Act).
- Quality Assurance Manager who is responsible for quality control (he/she must have 3 years or more experience in quality assurance work and meet other requirements under GQP).
- Safety Control Manager who is responsible for safety assurance (he/she must have 3 years or more experience in safety control work and meet other requirements under GVP).
[*3 Ministerial Ordinance on Standards for Quality Control for Drugs, Quasi-drugs, Cosmetics and Regenerative Medicine Products.]
[*4 Ministerial Ordinance on Standards for Post-Marketing Safety Management of Pharmaceuticals, Quasi-Drugs, Cosmetics, Medical Devices, and Regenerative Medicine Products.]
What is a “Manufacturing License” and Its Requirements?
A manufacturing license (or registration or accreditation (for foreign manufacturers), as the case may be) is a business license required for manufacturing Pharmaceuticals. Here, “manufacturing” refers to the entire manufacturing process, including packaging, affixing statutory labeling, and storage by the MAH, prior to shipment approval. A manufacturing license must be obtained for each manufacturing site according to the types of Pharmaceuticals and manufacturing process to be conducted at the manufacturing sites. If an MAH actually manufactures the Pharmaceuticals by itself, it needs to obtain a manufacturing license in addition to a marketing business license. On the other hand, if an MAH does not manufacture the Pharmaceuticals by itself and contracts with contract manufacturers to manufacture the Pharmaceuticals, the MAH does not need to obtain a manufacturing license by itself.
The major high-level requirements for a manufacturing license are as follows:
- The license applicant does not fall under any disqualification prescribed under the PMD Act.
- The structure and equipment of the manufacturing sites comply with the standards specified by the relevant MHLW ordinance.
- A manufacturing supervisor is appointed (who must be a pharmacist, in principle).
What Licenses Are Required to Sell Pharmaceuticals?
In order to sell Pharmaceuticals in Japan, the seller must hold the relevant license depending on the type of sales.
The type of sales are as follows:
- Pharmacy: A pharmacy may sell or provide Pharmacy-only Drugs and OTC Drugs. “Pharmacy-only Drugs” means Pharmaceuticals other than OTC Drugs, such as ethical drugs.
- Store-based distribution: A store-based distribution license holder may sell or provide OTC Drugs at a store.
- Household distribution: A household distribution license holder may sell or provide, via household distribution, General OTC Drugs that meet standards specified by the Minister, such as being resistant to deterioration over time. Household distribution is a method of sale in which Pharmaceuticals are left with consumers (e.g., general households) in advance and the consumers will be charged a fee upon using them.
Wholesale distribution: A wholesale distribution license holder may sell or provide Pharmaceuticals to pharmacies, MAHs, manufacturers or sellers of Pharmaceuticals; or hospitals, clinics, clinics for domesticated animals and others specified by the relevant MHLW ordinance.
What is the Outline of Post-Marketing Pharmacovigilance?
The PMD Act provides for various post-marketing pharmacovigilance measures to be taken by various parties including MAHs. The major post-marketing pharmacovigilance measures are as follows:
- Publication of precautions and other information: MAHs must publish precautions and other information of certain Pharmaceuticals (basically, ethical drugs) on the PMDA’s website (so-called “electronic package inserts”).
- Collection, review and provision of quality and safety information: MAHs, wholesalers or Special Foreign Approval Holders must make efforts to collect, review and provide information related to quality and safety and other information required for the appropriate use of Pharmaceuticals.
- Measures to prevent hazards: In the case of actual or potential health hazards, MAHs or Special Foreign Approval Holders must take the required measures for prevention of health hazards such as disposal, recall or suspension of sales of the Pharmaceuticals, and provision of required information.
- Adverse event reporting: MAHs or Special Foreign Approval Holders must report the adverse events specified by the relevant MHLW ordinance to the Minister in accordance with specified procedures under the MHLW ordinance. They must report to the Minister within the prescribed timeframe depending on the severity of the adverse events.
- Early post-marketing phase vigilance (EPPV): EPPV is required for new Pharmaceuticals in order to promote proper use of the Pharmaceuticals and to collect information related to safety. EPPV needs to be conducted for 6 months after the product launch and MAHs must make medical institutions aware that the Pharmaceuticals are subject to EPPV and request them to report to the MAHs any serious adverse events related to the Pharmaceuticals.
- Post-marketing surveillance (PMS): PMS are investigations or studies conducted after Pharmaceuticals are placed on the market to collect, detect, confirm or verify information on the quality, efficacy and safety of the Pharmaceuticals when safety monitoring is considered necessary. There are three types of PMS under GPSP*5 (i) use results surveys, (ii) post-marketing database surveys and (iii) post-marketing clinical trials. PMS is conducted for the majority of new Pharmaceuticals.
- Compliance with GVP: Methods of post-marketing safety control for Pharmaceuticals must comply with GVP as a condition for the marketing business license.
[*5 Ministerial Ordinance on Standards for the Conduct of Post-Marketing Surveys and Tests for Pharmaceuticals.]
What is “Re-Examination” of Pharmaceuticals?
Re-examination of Pharmaceuticals is conducted for new Pharmaceuticals placed on the market to re-examine the effects/efficacy, safety and quality etc. of such new Pharmaceuticals based on the data collected during the re-examination period. MAHs must conduct investigations concerning usage results and other matters specified by the relevant MHLW ordinance during the re-examination period, and report the results to the PMDA every year, in principle. Based on the results of the re-examination, one of the following will apply regarding the relevant Pharmaceutical:
- Cancellation of the marketing approval.
- Deletion or amendment of the stated effects/efficacy.
- No special measures to be taken (i.e., the marketing approval continues to be effective without changes).
The following are the re-examination periods for new Pharmaceuticals:
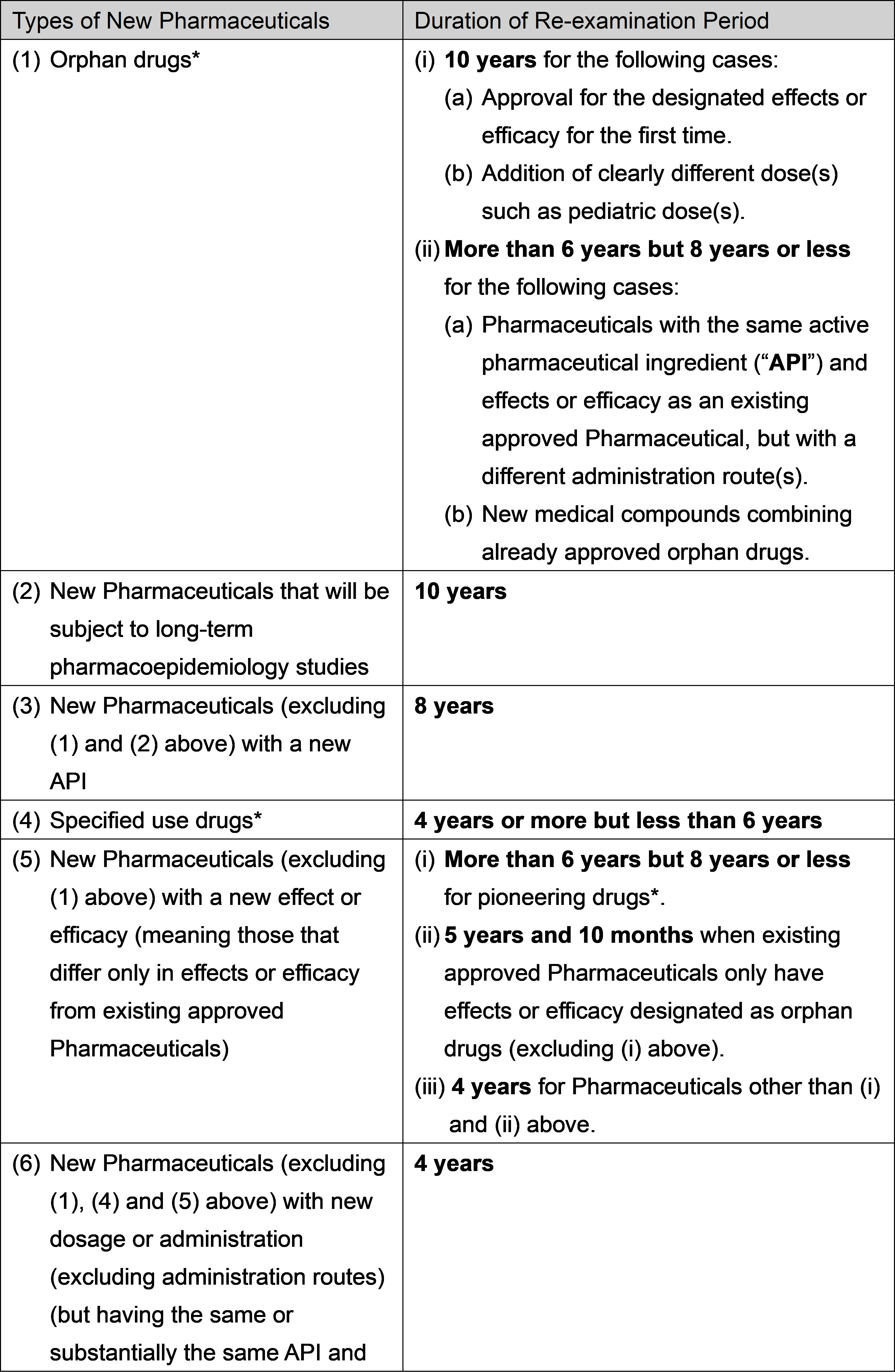
Reference: MHLW notice dated January 16, 2024
*Please see 2-12 for details of orphan drugs, specified use drugs and pioneering drugs
In addition, the re-examination period is said to sometimes operate as an exclusivity period for Pharmaceuticals. Please read more about such de-facto exclusivity effect of re-examination in the article below:
What is the Outline of Advertisement Regulations For Pharmaceuticals?
Advertisement of Pharmaceuticals is subject to strict advertisement regulations including under the PMD Act as well as the Standards for Appropriate Advertisement of Pharmaceuticals, etc. (“Appropriate Advertisement Standards”) and the Guidelines on Sales Information Provision Activities (Promotional Activities) for Ethical Drugs (“Promotional Activity Guidelines”) - both issued by the MHLW.
The major advertisement restrictions for Pharmaceuticals are as follows:

Furthermore, industrial associations have their own self-regulatory standards. For example, the Japan Pharmaceutical Manufacturing Association (JPMA), which is mainly for MAHs of ethical drugs, has self-regulatory standards such as the Guidelines for the Preparation of Ethical Drug Product Information Summaries and Related Materials (“Preparation Guideline”), and the Code of Practice and Transparency Guidelines. The Preparation Guideline in particular, provides very detailed standards for promotional materials.
Is There Any Restriction on Providing Benefits to HCPs?
According to the notice “Restriction on the Provision of Premiums in the Ethical Drug, Medical Device and Clinical Laboratory Industries” (“Notice”), issued under the Act against Unjustifiable Premiums and Misleading Representations, those engaging in manufacturing or selling business of ethical drugs or medical devices, or clinical laboratory business must not provide medical institutions and other similar institutions, including but not limited to, hospitals, clinics, pharmacies or healthcare professionals and employees belonging to these institutions (collectively, “Medical Institutions”), with premiums beyond the scope of the goods or services necessary for use of the ethical drugs or medical devices or utilization of clinical laboratories as well as any other scope deemed appropriate in light of normal commercial practices, as a means to unjustifiably induce transactions for ethical drugs, medical devices or with clinical laboratories. In other words, MAHs and sellers of ethical drugs, etc. cannot provide unjustifiable premiums to the Medical Institutions.
There are no guidelines for the Notice. In this regard, the following Fair Trade Councils have their own Fair Competition Codes and Enforcement Rules of the Fair Competition Codes (collectively, “Fair Competition Codes”) that are approved by the Consumer Affairs Agency and the Japan Fair Trade Commission:
- Fair Trade Council of the Ethical Pharmaceutical Drugs Marketing Industry
- Fair Trade Council on Ethical Drug Wholesaling
- Japan Fair Trade Council of the Medical Devices Industry
- Fair Trade Council of the Clinical Laboratory Industry
These Fair Competition Codes and related guidelines elaborate the standards for determining whether certain provision of premiums to the Medical Institutions is prohibited or not under the Notice. Although these Fair Competition Codes are only applicable to members of the relevant Fair Trade Councils, they are still useful for non-member companies and as guidance to interpret the Notice, given that there are no other guidelines for the Notice.
Are There Any Fast-Track Programs For Approval For Pharmaceuticals?
There are some programs that would accelerate the marketing approval process in Japan such as the “Priority Review Program”. The marketing approval review process for the Pharmaceuticals subject to the Priority Review Program may be prioritized over that for other Pharmaceuticals.
The Priority Review Program is available for the following Pharmaceuticals:
What’s Next?
In our next post, we will provide a detailed overview of the regulations regarding “Medical Devices”. Stay tuned!
Previous Article
Healthcare & Pharmaceutical Regulations in Japan
Click here to view the article in PDF format.
Member





SHARE![]()
![]()
![]()